
What is Gene Editing?
Gene editing is the process of intentionally modifying an organism’s genome through the insertion, deletion, or replacement of DNA.
Editing is dependent upon creating a double-strand break (DSB) at a particular point within the genome. This is accomplished with engineered nucleases that are targeted to specific genomic loci with guide molecules, or with sequence specifications programmed into the nuclease itself.
Methods of gene editing include: CRISPR/Cas9, Cas12, ZFNs, TALENs, and meganucleases. Each of these systems operate by targeting an engineered nuclease to an exact location within the genome where they bind and create sequence-specific DSBs.
A target DNA sequence can be deleted, modified or replaced using the cell’s endogenous repair machinery. Insertions and deletions at the edit site can range in size from a large sequence to a single base pair. The example above provides a general representation of a sequence addition and a sequence deletion; however, many more editing variations are possible.
Nuclease engineering, optimized delivery conditions and cellular repair mechanisms enable researchers to manipulate segments of DNA and the genes they encode for. You can learn more about the principles of gene editing and the various types of editing here.
Why Characterize Editing Outcomes?
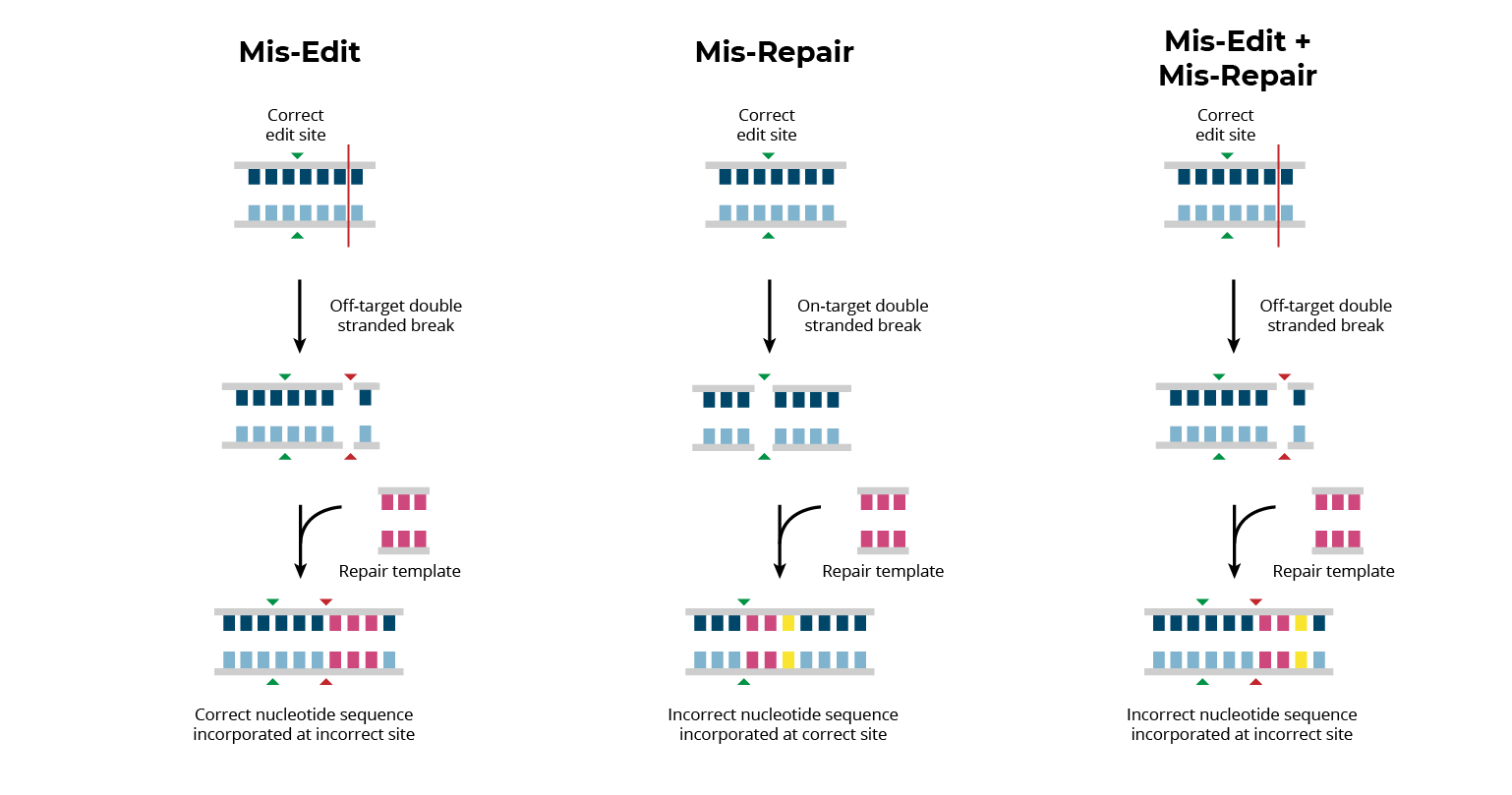
Editing associated errors, both on- and off-target, result in genomic variants which could impact patient safety. In order to realize the clinical potential of gene editing treatments, all editing associated errors must be identified and quantified. Editing-associated errors can be broadly classified into three categories: mis-edits, mis-repairs, and mis-edit/mis-repair combinations.
Mis-edits occur when the editing enzyme creates off-target DSBs at homologous or random sites in the genome. Mis-edits typically result in small insertions or deletions (indels) of nucleotides at unintended genomic loci.
Mis-repairs occur when a cell’s endogenous machinery incorrectly repairs on-target nuclease-induced DSBs. Mis-repairs result in unintended changes to the edit site that can vary from single base pair insertions/deletions to large genomic rearrangements.
Additionally, combinations of these errors can take place in which a mis-repair occurs at an off-target site. While less frequent, this can result in genomic changes that are particularly complex and difficult to identify.

All editing-associated errors can result in genomic variants that are potentially harmful and represent risk for the patient. Measuring nuclease-induced changes at the edit site and throughout the genome is necessary, since it is possible for even low-frequency, heterogeneous, rearrangements to have serious consequences. Understanding the existing heterogeneity and spontaneous rate or rearrangements that exist pre-editing is essential for measuring editing effects.
Where Does dGH Fit In?
directional Genomic Hybridization (dGH) is an efficient and practical technique for measuring genomic baselines, on- and off-target editing effects and the persistence of genomic variants over time.
By measuring structural variation in many single cells, dGH can be used to quantitate individual on- and off-target variants, including those that are present in less than one percent of the edited cells.
dGH utilizes an innovative chromosome preparation technique, that incorporates analog nucleotides, daughter strand degradation and synthetic TAG-oligos. This design enables single assay de novo identification of sequence, location and orientation for each genomic structural change.
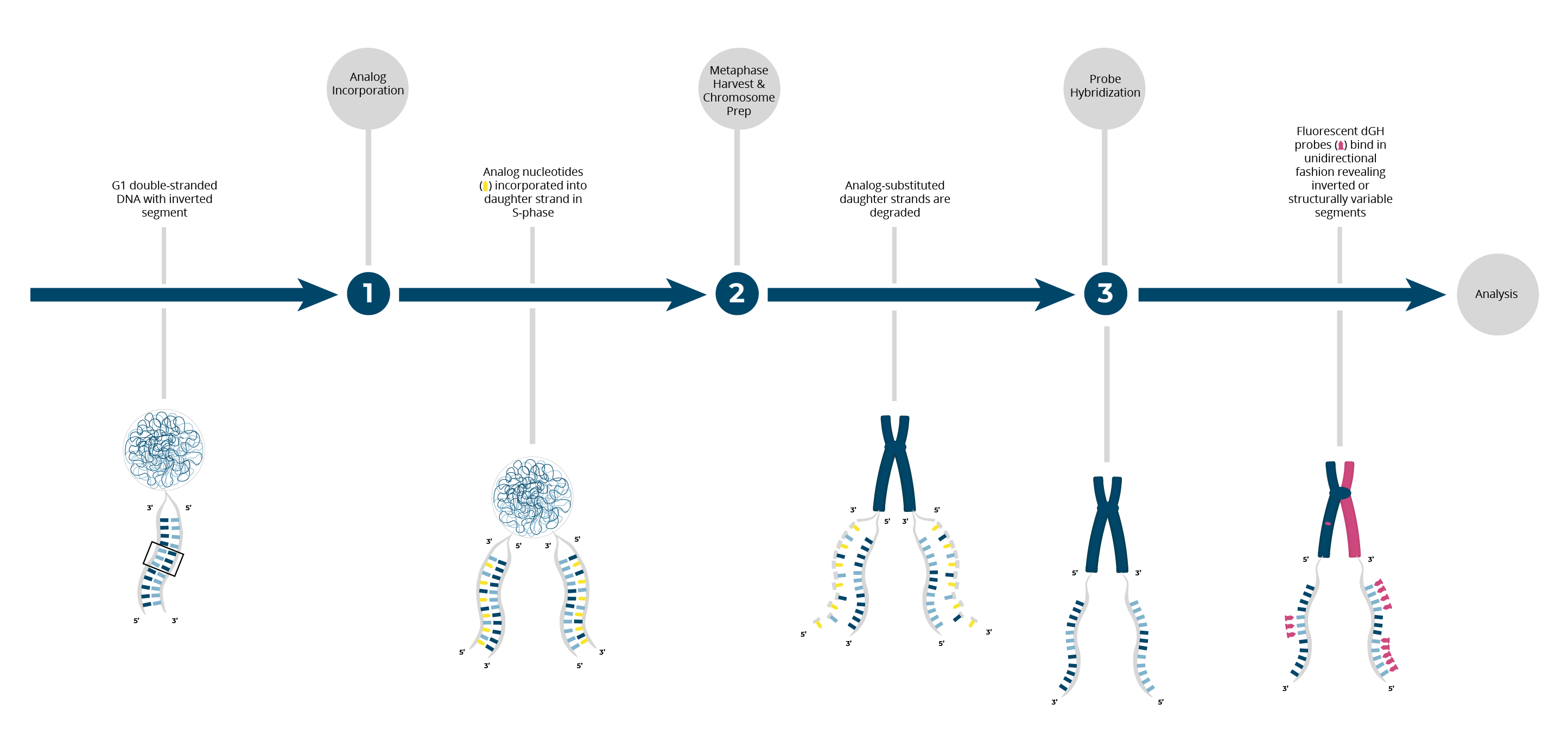
KromaTiD’s synthetic TAG-oligos are bioinformatically designed to be repeat-free. dGH TAG-oligos bind single-stranded DNA in a unidirectional manner, allowing for the de novo visualization of complex structural rearrangements which are undetectable with any other method. You can learn more about dGH and how it works here. 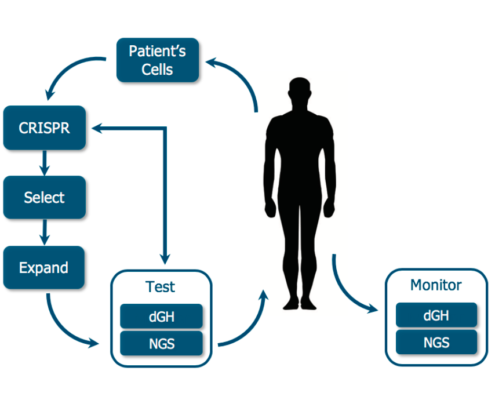
dGH provides a straightforward method for obtaining quantifiable structural genomic data at the single cell level. dGH fits into the gene editing workflow as an orthogonal and complementary analytical technique to sequencing.
dGH can be employed at multiple time points to monitor chromosomal structure throughout the lifetime of the editing process.
Pre-edit: dGH provides a measurement of pre-existing structural variation which is unique to each model, person and cell type. In addition to establishing a genomic baseline, dGH can be used for process optimization. dGH can use the on-and off-target rates of structural variation to monitor the effects of differing process variables including engineered-nuclease variants, guide strand choice and delivery method, and direct comparisons within the cell lines of interest.
Post-edit: dGH can measure structural rearrangements at multiple time points immediately following editing. In addition, dGH can be used to track the persistence of measured variants over time. From establishing a baseline structural variation rate, to measuring editing-associated changes, to tracking genomic integrity after treatment, dGH provides a comprehensive structural genomic profiling technique which helps to ensure that any genomic rearrangements resulting from editing are accounted for.
How is dGH Different?
In contrast to the many sequencing-based analysis approaches available, dGH provides direct visualization of structural variants at the single cell level. Unlike sequencing, dGH does not rely on pooled DNA and it provides rearrangement data within the context of the cell.

Fundamental differences between dGH and sequencing result in fundamental differences between the data that each platform provides.
Sequencing based approaches inherently provide data based on isolated and pooled DNA from edited cell populations. This means that the cellular context in which genetic aberrations occur is entirely lost. The dGH process, however, gathers data from actively dividing cells, resulting in a cellular ‘snapshot’ of the genetic structural variation that has taken place within that cell up until the point of arrest and fixation. 
Additionally, sequencing approaches rely on PCR amplification of a specific genomic region of interest. This type of amplification subjects results to PCR biases and makes de novo analysis and analysis of regions high in repeats challenging.
Ultimately sequencing is ideal for high throughput analysis of specific regions of the genome, while dGH can provide whole genome, de novo characterization of low-frequency or heterogeneous variants. Each method has different strengths that provide unique data and both methods are required in order to gain a comprehensive understanding of editing and its associated risks.
Gene Editing Services and Custom Assay Development
To solve your gene editing analysis problems, we offer both custom dGH assay development solutions, and full analysis services for all gene editing applications:

Measure the level of structural variation in your starting cell population in order to establish your pre-edit baseline rate.
Perform de novo tracking of edited chromosomes in order to get a profile of chromosome specific, edit-induced, and random structural variation.

Discover and quantify low-frequency structural rearrangements within heterogeneous cell populations

Get quantitative and qualitative data regarding the consistency and prevalence of structural on/off-target effects at the single cell level.
KromaTiD offers de novo or targeted genomic profiling of editing associated structural errors for each system and each batch. dGH can give you a quantitative assessment of structural rearrangements at the single-cell level which, when combined with NGS or Sanger sequencing, provides a regulator-ready data package for clinical trials and therapeutic gene editing.