
On a cellular level, the behavior of biological systems is guided by nucleic acid molecules, such as DNA. Cells lacking a gene for a given protein cannot manufacture it, unless it is supplied by some exogenous source like a virus. However, a significant gap exists between a cell harboring a specific DNA sequence and its ability to manufacture the corresponding protein. Various barriers can intervene between genotype and phenotype. Scientists have by now conceived of a dizzying array of methods and devices with which to read the sequence of virtually any molecule that contains one. Here, we will explore two such platforms which enable researchers to scrutinize the intermediate stage between genes and proteins: Microarray and RNA-Seq, two of the most widely employed methods in modern transcriptomics.
The term, “microarray” refers to the array of probe spots covalently bound to a substrate upon which probe binding takes place. Each spot may be only a micron in size and the probes may be nucleic acid, carbohydrate or peptide-based. The substrate to which those molecules are attached is typically a glass slide, or “chip.” However, alternatives include beads, and substrates can be made from silica, polystyrene, or other plastics.1,2
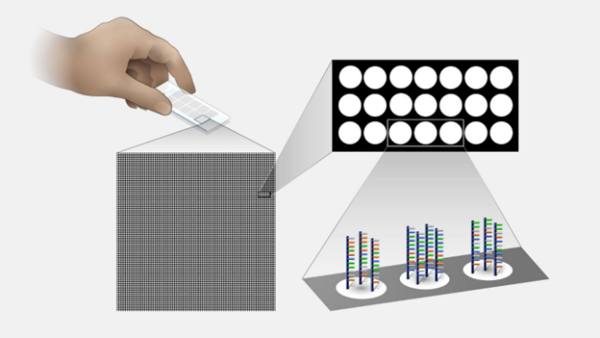 A close-up view of a microarray slide. (Source: Smith, M; 2023)
A close-up view of a microarray slide. (Source: Smith, M; 2023)
Laboratories have two options for obtaining the necessary materials to run microarray, each with advantages and disadvantages – users may buy supplies from large-scale manufacturers, or they may manufacture them in house themselves. A laboratory with a smaller budget that doesn’t expect a very high sample volume may opt to purchase spot-printing equipment. Though the proportion of spots which bind targets effectively may not be as high as with more costly, pre-printed products, one very valuable benefit is that owning the spot printer allows a user to customize the probe set. Data generated can then be catered to specific scientific questions for which there may not be ideal pre-printed product options on the market.1
However, momentum seems to be moving away from microarray in favor of RNA-Seq because there is a key limiting factor in microarray which it shares with other hybridization-based technologies. Microarray can only gather data on sample molecules which can be bound by the probes being used. RNA-Seq, though significantly more costly than microarray, requires less upstream effort in order to assay targets such as non-coding RNA, single-nucleotide polymorphisms, fusion genes and to map exon junctions. Microarrays can measure these, but bioinformatics work must take place in advance in order to create the specialized chips required. In addition, RNA-Seq will detect splice variants and novel sequences, which microarray can only measure if a user somehow has enough knowledge of these in advance to create and include the necessary probes on the chip.3
Nevertheless, both techniques are vulnerable to their own blind spots, which is important to understand in order to consider how best to validate data with an orthogonal platform. The upstream effort mentioned with respect to RNA-Seq refers to steps required in order to mitigate a bias in favor of larger target molecules. RNA-Seq sample RNA molecules must be fragmented in preparation for sequencing. Longer molecules produce more fragments and thus, more target molecules available to sequence.3 Detection of certain categories of targets thus requires physical screening for smaller molecules at the outset. Likewise, microarray is plagued by an Achille’s heel of its own in that statistical variation in the sequences of the target molecules result in an unpredictable percentage of total base pairs being Guanine-Cytosine (GC) bonds. GC base pairs link together with three hydrogen bonds, whereas Adenine-Thymine (AT) pairs share only two. This unpredictable overall difference in bond strength from target to target reduces the proportion of total probe-target molecule pairs that will remain successfully hybridized at the end of the process, diminishing the value of the final data set.
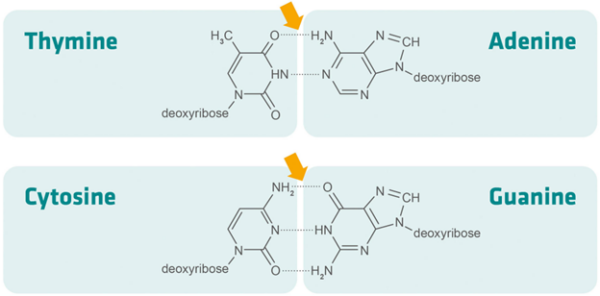
Comparison of molecular structure between AT and GC base pairs, with arrows indicating hydrogen bonds. (Source: Mészáros, E; 2022)
The types of techniques which, for a time are considered industry-standard, legacy, or state-of-the-art are forever changing within the broader field of biotechnology. RNA-Seq appears to be on its way to becoming a mainstay platform. Yet it is important to keep an eye on the horizon for new methods and for innovative scientists who may repurpose or modify older methods, perhaps enhancing them through use of new bioinformatics tools and/or artificial intelligence algorithms.
References
- Terra Universal. (2015, December 30). Microarray Platforms: Beads, Slides or Chips? Laboratory-Equipment.com https://www.terrauniversal.com/blog/microarray-platforms-beads-slides-or-chips#:~:text=Microarray%20analysis%20(control%20%3D%20red),easily%20done%20using%20affordable%20instrumentation.
- Smith, Mike. (2023, August 9). MICROARRAY TECHNOLOGY. National Human Genome Research Institute. https://www.genome.gov/genetics-glossary/Microarray-Technology
- Mantione, K. J., Kream, R. M., Kuzelova, H., Ptacek, R., Raboch, J., Samuel, J. M., & Stefano, G. B. (2014). Comparing bioinformatic gene expression profiling methods: microarray and RNA-Seq. Medical science monitor basic research, 20, 138–142. https://doi.org/10.12659/MSMBR.892101
- Mészáros, E. (2022, March 24). How to design primers for PCR. INTEGRA. https://www.integra-biosciences.com/china/en/blog/article/how-design-primers-pcr